Integrating Multi-Omics Data to Enhance Protein-Protein Interaction Predictions Using Variational Graph Autoencoders
Table of Contents
Highlights
Protein-protein interaction (PPI) networks are essential for understanding molecular mechanisms in biology, with applications in drug discovery, functional genomics, and disease pathway identification. However, existing computational methods often rely on static interaction datasets, lack multi-omics integration, and require high computational resources.
To address these limitations, we present PPI-OMEGA (Protein-Protein Interaction with Omics-Enhanced Graph Autoencoder), a Variational Graph Autoencoder (VGAE)-based framework that integrates RNA expression and immunohistochemistry (IHC) protein expression data to enhance PPI prediction. Our model generates biologically meaningful protein embeddings by leveraging graph convolutional layers alongside multi-omics features.
🟢 Key Findings:
- Multi-omics integration significantly improves predictive accuracy: The combined RNA and protein expression model achieved the highest AUROC (0.9235) and AP (0.9318), outperforming RNA-only and protein-only models.
- Protein expression contributes more than RNA expression: The model using only IHC protein expression performed nearly as well as the multi-omics model (AUROC = 0.9215, AP = 0.9297), while the RNA-only model had a more significant drop in performance.
- Multi-omics features enhance biological relevance: The learned protein embeddings reflect functional characteristics, successfully distinguishing between housekeeping and context-dependent proteins.
- Graph-based embeddings improve model generalizability: The PPI-OMEGA model captures structured protein-protein interaction patterns, reducing computational overhead while enabling potential downstream disease association studies and functional classification.
This research highlights the power of graph-based multi-omics integration for improving disease-relevant PPI modeling and advancing precision medicine applications. Some future improvements include integrating protein sequence data and incorporating attention mechanisms to further refine interaction predictions.
Introduction
Protein-protein interactions (PPIs) are fundamental to cellular function, governing key processes such as signal transduction, gene regulation, and metabolic pathways. Understanding these interactions is crucial for identifying disease mechanisms, discovering therapeutic targets, and advancing functional genomics.
Traditional computational models for PPI prediction primarily rely on static datasets derived from high-throughput experiments. However, these models face critical limitations, including their inability to incorporate dynamic biological contexts, lack of multi-omics integration, and high computational costs when applied to large-scale datasets. As a result, they often fail to capture tissue-specific interactions and may overlook biologically significant but previously uncharacterized PPIs.
PPI-OMEGA addresses these challenges by integrating multi-omics data into a Variational Graph Autoencoder (VGAE) framework. Unlike conventional methods, our approach leverages both RNA expression profiles and immunohistochemistry (IHC) protein expression data to refine PPI predictions. By embedding multi-omics features within a graph-based learning architecture, PPI-OMEGA generates biologically meaningful representations of protein interactions.
This study details our methodology, including data preprocessing, model architecture, and evaluation metrics. Through an ablation study, we demonstrate that multi-omics integration significantly improves prediction accuracy, with the combined RNA and protein expression model achieving the highest AUROC (0.9235) and AP (0.9318). We also analyze the biological relevance of our learned protein embeddings, revealing their ability to distinguish housekeeping and context-dependent proteins. Finally, we discuss the broader implications of our findings, particularly in precision medicine, disease pathway analysis, and functional classification.
Methods
🟢 OverviewThe PPI-OMEGA framework is designed to enhance protein-protein interaction (PPI) predictions by integrating multi-omics data into a graph-based learning model. We leverage a Variational Graph Autoencoder (VGAE) to capture complex dependencies in PPI networks, using RNA expression and immunohistochemistry (IHC) protein expression as node attributes.
🟢 Dataset & PreprocessingWe constructed our PPI dataset using the STRING database, which contains interaction confidence scores for protein pairs. The dataset was filtered to retain the top 5% high-confidence interactions and mapped to human gene identifiers.
Additional multi-omics features were integrated from The Human Protein Atlas:
- RNA Expression Data: Processed from 35 tissue types, normalized using TPM values.
- IHC Protein Expression Data: Derived from 45 tissues, discretized into 4 bins of different expression levels.
Each protein was represented as a node, and edges were formed based on STRING database interactions. Node attributes included:
- Structural Features: Encoded from known PPI networks.
- Node Feature Data: Preprocessed RNA and/or IHC protein values as needed.
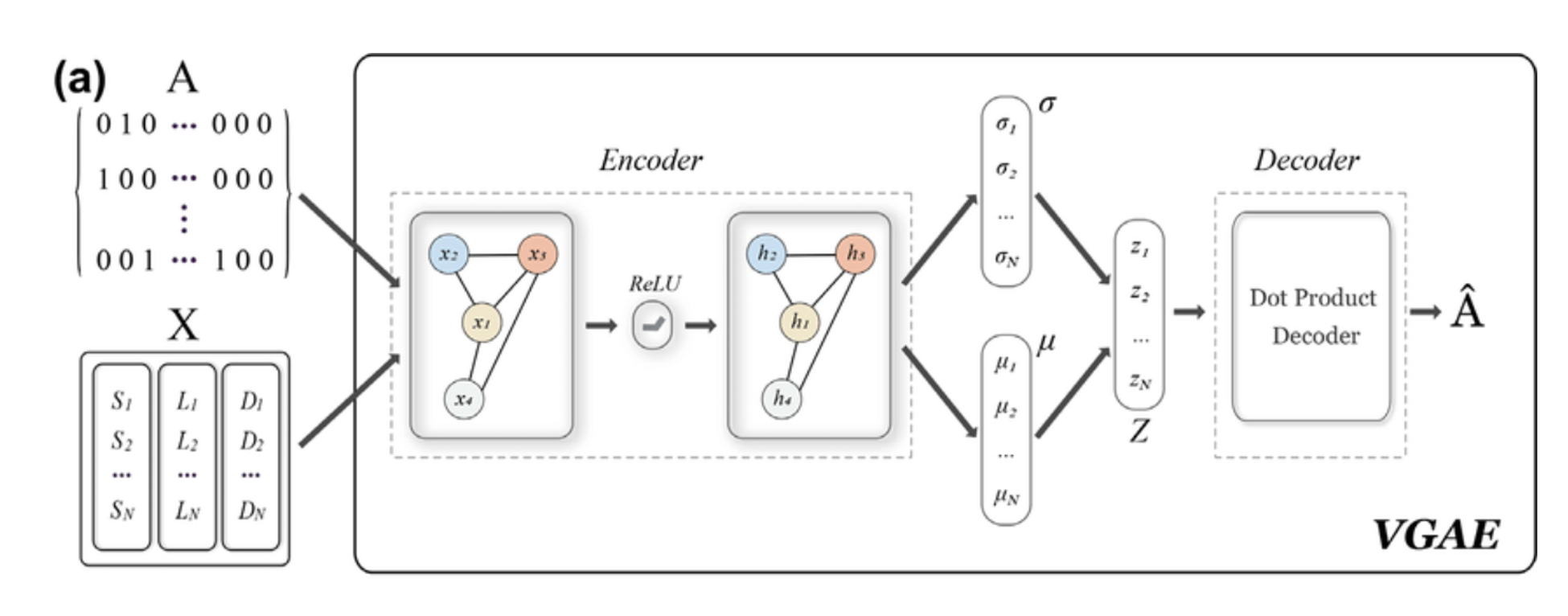
Our model follows a standard VGAE framework:
- Encoder: Two-layer Graph Convolutional Network (GCN) extracts node embeddings.
- Latent Space: A probabilistic layer learns a Gaussian distribution for protein representations.
- Decoder: Dot product of embeddings reconstructs the adjacency matrix.
To optimize model performance, we performed a grid search over key hyperparameters, including the dropout rate, learning rate, and weight decay. Each configuration was evaluated on the validation set, selecting the combination that yielded the highest AUROC and AP scores.
- Dropout Rate (p): {0.3, 0.4, 0.5}
- Learning Rate (α): {0.001, 0.005, 0.01}
- Weight Decay (λ): {5e-4, 1e-3, 5e-3}
The best-performing configuration was found to be:
- Dropout Rate: 0.3
- Learning Rate: 0.01
- Weight Decay: 5e-4
This configuration resulted in the highest AUROC (0.9235) and AP (0.9318), significantly improving predictive accuracy compared to suboptimal hyperparameter choices.
To ensure robustness, we implemented an early stopping strategy based on validation AUROC. If no improvement of at least 0.01 was observed for 60 consecutive epochs, training was halted to prevent overfitting. Additionally, dropout regularization was applied to mitigate overfitting while ensuring stable convergence.
🟢 Training & EvaluationThe model is trained using a binary cross-entropy loss with a Kullback-Leibler (KL) divergence regularization term: \(\mathcal{L} = \mathcal{L}_{recon} + \beta \mathcal{L}_{KL}\). We used the Adam optimizer with a learning rate of 0.001. Training was stopped when validation loss did not improve by a minimum threshold within a set number of epochs.
🟢 Ablation StudyTo assess the impact of multi-omics data integration, we conducted an ablation study during which we trained and evaluated the model under four different feature input conditions:
| Feature Set | Description |
|---|---|
| No Features | Only the network structure (adjacency matrix) was used as input. |
| RNA Only | RNA expression profiles were included as node attributes. |
| Protein Only | IHC protein expression levels were included as node attributes. |
| Combined (RNA + Protein) | Both RNA expression and IHC protein expression features were integrated. |
The performance of each model variant was evaluated using AUROC and AP (Average Precision) scores.
Read more about our method architecture and training details on our report.
Results
To evaluate the impact of different input feature combinations on model performance, we conducted an ablation study. Table 2 presents AUROC and AP scores for four configurations: using no features, using only RNA expression, using only protein expression, and combining both features.
| RNA Exp. | IHC Protein Exp. | AUROC | AP |
|---|---|---|---|
| ❌ | ❌ | 0.8194 | 0.8280 |
| ✅ | ❌ | 0.8888 | 0.9026 |
| ❌ | ✅ | 0.9215 | 0.9297 |
| ✅ | ✅ | 0.9235 | 0.9318 |
Our results demonstrate that integrating both RNA and protein expression features led to the highest performance, with an AUROC of 0.9235 and an AP of 0.9318. This confirms that incorporating multi-omics data enhances PPI prediction compared to using RNA or protein expression alone.
Notably, the model trained with protein expression (IHC data) achieved an AUROC of 0.9215 and an AP of 0.9297, surpassing the RNA-only model (AUROC = 0.8888, AP = 0.9026). This suggests that protein expression features provide a stronger signal for identifying functional interactions. Meanwhile, the addition of RNA expression demonstrates diminishing return, which highlights needs to look for potential more relevant data.
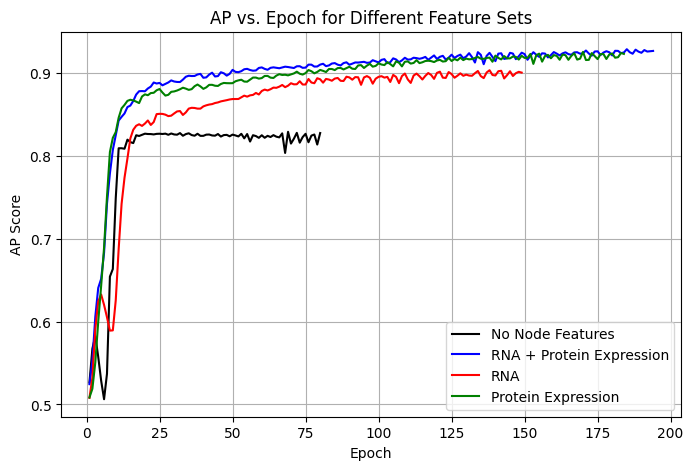
🟢 Training DynamicsTo evaluate model convergence and performance stability, we tracked training dynamics by monitoring AP (Average Precision) scores across epochs (Figure 3). The multi-omics model, which integrates both RNA and protein expression features, demonstrated faster convergence and higher final AP scores compared to single-feature models.
The results indicate that:
- The multi-omics model achieves the highest AP score and converges faster than RNA-only and protein-only models.
- The protein-only model performs almost as well as the multi-omics model, reinforcing the importance of protein expression features in PPI prediction.
- The RNA-only model lags in both convergence speed and final AP, suggesting that RNA expression alone provides limited predictive power.
- The model trained without node features struggles with stability and achieves the lowest AP score, confirming that incorporating biological features significantly improves PPI predictions.
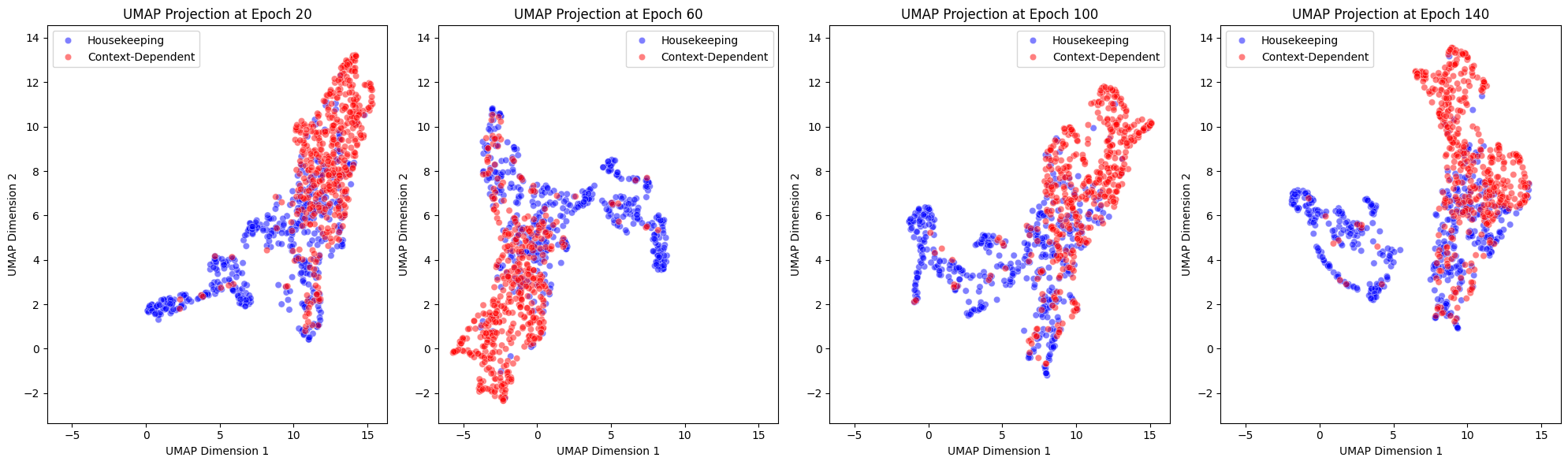
To assess the biological relevance of the learned protein embeddings, we examined how the latent representations evolved during training. The model successfully captures biologically meaningful relationships, distinguishing between housekeeping and context-dependent proteins and structuring embeddings based on network connectivity.
As shown in Figure 4, we visualized the UMAP projections of protein embeddings at different training epochs, classifying proteins as either housekeeping or context-dependent based on functional annotations. The results show that:
- The separation between housekeeping and context-dependent proteins increases over training, indicating that the model progressively refines biologically meaningful representations.
- Some overlap remains, likely reflecting functional interactions between proteins that span both categories.
To further investigate how network structure influences embedding formation, we analyzed the variance of the latent representation mean (μ) for highly connected versus sparsely connected proteins (Figure 5). The findings suggest that:
- Highly connected proteins converge faster, with their embeddings stabilizing early in training due to strong structural constraints.
- Sparsely connected proteins exhibit greater variance fluctuations before stabilizing, requiring more training epochs to reach a meaningful latent space representation.
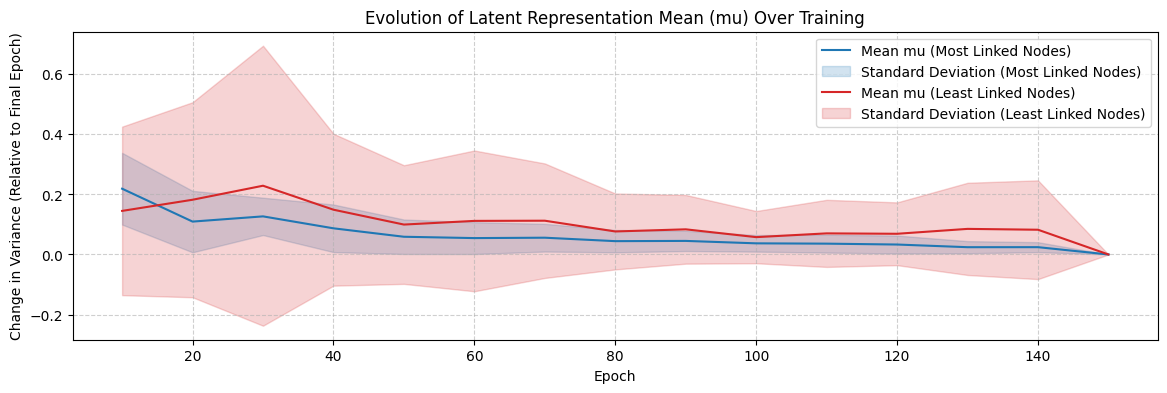
Read more detailed results and interpretation on our report.
Discussion & Future Work
🟢 Enhancing PPI Prediction with Multi-OmicsPredicting protein-protein interactions (PPIs) is essential for understanding biological systems, disease mechanisms, and drug discovery. Traditional models primarily rely on sequence data, but our study demonstrates that incorporating RNA and protein expression significantly improves prediction accuracy. This multi-omics approach captures functional relationships beyond what sequence data alone can reveal. Future work will include integrating protein sequence features alongside multiomics data to build a more comprehensive and accurate PPI prediction framework.
🟢 Handling Dropped or Unused DataPreprocessing decisions impact the quality of PPI predictions. While thresholding and PCA reduce noise, they may also remove informative low-confidence PPIs or rare expression patterns. To improve data retention and maintain biological signal integrity, future work will explore:
- Adaptive thresholding to refine data inclusion dynamically.
- Data imputation to recover missing but meaningful information.
- Feature selection to balance dimensionality reduction and biological relevance.
Our reliance on immunohistochemistry (IHC) data introduces variability in protein quantification. More precise techniques will enhance the robustness of our model, including:
- Using mass spectrometry-based proteomics for accurate protein quantification.
- Normalizing RNA-protein expression values across datasets.
- Incorporating time-series expression data to capture dynamic PPI interactions.
- Exploring single-cell transcriptomics to improve resolution in cellular studies.
While our study focused on multi-omics data, protein sequence information remains a fundamental component of PPI prediction. Many models leverage structural motifs and conserved domains for interaction analysis. Future work will integrate sequence-based features with multi-omics data to enhance accuracy and uncover novel PPIs relevant to disease research.
🟢 Advancing Model ArchitectureImproving model interpretability and predictive power requires architectural refinements. Planned enhancements include:
- Implementing attention mechanisms to prioritize biologically significant interactions.
- Expanding multi-omics integration to include epigenetic and metabolic profiles.
- Addressing class imbalance to minimize false positives in real-world applications.
- Enhancing generalizability across tissues for precision medicine applications.
- Exploring graph-based deep learning to model complex biological networks.
More accurate PPI prediction models have broad applications in biomedical research. Our findings contribute to:
- Drug discovery – Identifying novel drug targets and reducing off-target effects.
- Disease modeling – Mapping interaction networks in genetic disorders and cancers.
- Precision medicine – Personalizing treatments based on molecular profiles.
- Systems biology – Providing deeper insights into cellular mechanisms.
Moving forward, we will expand our datasets, refine our methodologies, and collaborate with experimental biologists to validate findings. By bridging computational predictions with experimental validation, we aim to create a biologically informed AI-driven tool for PPI analysis, driving new discoveries in molecular biology and therapeutic research.